User guide
All options
Getting started with pycrispr
pycrispr requires a YAML file (experiment.yaml) that contains information of the experiment, and available CRISPR sgRNA libraries:
rename: #rename to .fq.gz
L8_S1_L001_R1_001.fastq.gz: L8.fq.gz
S8_S3_L001_R1_001.fastq.gz: S8.fq.gz
S15_S4_L001_R1_001.fastq.gz: S15.fq.gz
library: kinase
lib_info:
kinase:
index: /home/user/Documents/references/index/hisat2/dub-only/kinase.index
fasta: /home/user/Documents/references/fasta/Human/kinase/kinase.fasta
sg_length: 20 #length of (shortest) gRNAs in library
species: hsa #human,hsa;mouse,mmu
left_trim: 0 #number of nucleotides to trim on 5' end of reads
bassik:
index: /home/user/Documents/references/index/hisat2/bassik/bassik-index
fasta: /home/user/Documents/references/fasta/Human/Bassik-library/bassik_lib.fasta
sg_length: 17
species: hsa
left_trim: 1 #number of nucleotides to trim on 5' end of reads
mismatch: 0 #mismatches allowed during alignment
stats:
bagel2_dir: /home/user/Documents/software/bagel2
type: mageck
comparisons: #test vs control (only alpha numerical characters allowed, except comma)
1: S8_vs_L8 #sample names are file names without extension
2: S15_vs_L8
3: S8,S15_vs_L8 #samples can be pooled
resources:
account: XXX
partition: cclake
max_jobs: 100 #maximum number of parallel jobs
trim:
cpu: 4
time: 60 #in minutes
fastqc:
cpu: 4
time: 60
count:
cpu: 8
time: 120
stats:
cpu: 1
time: 60
Note
You can delete/ignore the rename section if you do not need to rename your files, but please keep in mind that the sample names will be taken from the read file names by removing the file extension, which should be fq.gz. Also, the comparisons in the stats section should match this.
Preparing CRISPR-Cas9 screen data
Before running pycrispr, an analysis directory has to be created (can be any name or location), and should contain a sub-directory called reads. This sub-directory contains all the fastq files of your CRISPR-Cas9 screen experiment:
analysis_dir
├── reads
| ├── L8_S1_L001_R1_001.fastq.gz
| ├── S8_S3_L001_R1_001.fastq.gz
| └── S15_S4_L001_R1_001.fastq.gz
└── experiment.yaml
Important
Please note that pycrispr only accepts single-end NGS data, so if your data was sequenced in a paired-end fashion, only include the read mate that contains the gRNA sequence information (most commonly read 1). It also assumes that the first nucleotide sequenced is the first nulceotide of the gRNA sequence. If this is not the case, then the first n nucleotides can be skipped by setting left_trim to n in experiment.yaml.
Initiating the pipeline
To start the analysis, run:
$ pycrispr analysis -t 24
This will first rename the files according to experiment.yaml, use a total of 24 CPU threads, select the dub-only gRNA library, and use MAGeCK for pair-wise comparisons specified in experiment.yaml.
Output files
Multiple output files will be generated:
analysis_dir
├── count
| ├── alignment-rates.pdf
| ├── counts-aggregated.tsv
| ├── L8.guidecounts.txt
| ├── S15.guidecounts.txt
| ├── S8.guidecounts.txt
| └── sequence-coverage.pdf
├── envs
| ├── count.yaml
| ├── flute.yaml
| ├── join.yaml
| ├── mageck.yaml
| └── trim.yaml
├── logs
| ├── count
| ├── fastqc
| ├── mageck
| ├── multiqc
| └── trim
├── mageck
| └── many files
├── mageck_flute
| └── many files
├── qc
├── reads
| ├── L8.fq.gz
| ├── S8.fq.gz
| └── S15.fq.gz
├── scripts
| └── flute.R
├── dag.pdf
├── experiment.yaml
├── snakefile
└── utils.py
pycrispr will first create a Directed acyclic graph (DAG) for the current workflow.

Directed acyclic graph (DAG) for workflow
Graphs showing the alignment rates and the fold sequence coverage can be found in the count directory.
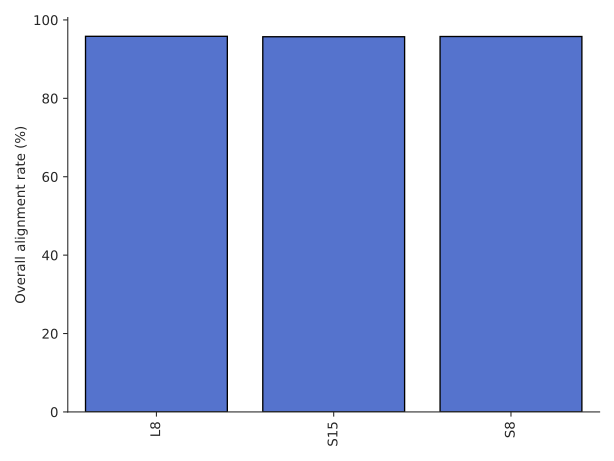
Alignment rates for each sample

Fold sequence coverage for each sample (number of aligned reads divided by number of gRNAs in library)
Report
After the analysis has finished, a HTML report can be generated (pycrispr-report.html):
$ pycrispr report
This report will be located in the analysis directory.
snakemake HTML report